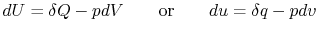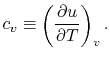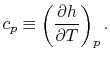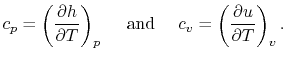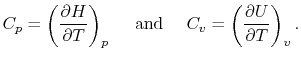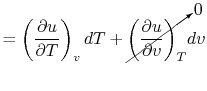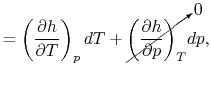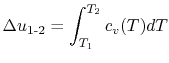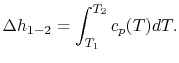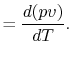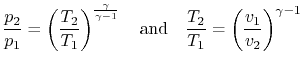Disusun Ulang Oleh:
Arip Nurahman
Pendidikan Fisika, FPMIPA. Universitas Pendidikan Indonesia
&
Follower Open Course Ware at MIT-Harvard University. Cambridge. USA.
Materi kuliah termodinamika ini disusun dari hasil perkuliahan di departemen fisika FPMIPA Universitas Pendidikan Indonesia dengan Dosen:
1. Bpk. Drs. Saeful Karim, M.Si.
2. Bpk. Insan Arif Hidayat, S.Pd., M.Si.
Dan dengan sumber bahan bacaan lebih lanjut dari :
Massachusetts Institute of Technology, Thermodynamics
Professor Z. S. Spakovszk, Ph.D.
Office: 31-265
Phone: 617-253-2196
Email: zolti@mit.edu
Aero-Astro Web: http://mit.edu/aeroastro/people/spakovszky
Gas Turbine Laboratory: home
2.4 Specific Heats: the relation between temperature change and heat
[VW, S& B: 5.6]
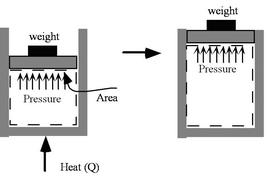
How much does a given amount of heat transfer change the temperature of a substance? It depends on the substance. In general
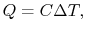 | (2..4) |
where
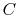
is a constant that depends on the substance. We can determine the constant for any substance if we know how much heat is transferred. Since heat is path dependent, however, we must specify the process, i.e., the path, to find
.
Two useful processes are constant pressure and constant volume, so we will consider these each in turn. We will call the specific heat at constant pressure 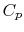 , and that at constant volume
, and that at constant volume 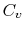 , or
, or 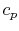 and
and 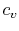 per unit mass.
per unit mass.
- The Specific Heat at Constant Volume
Remember that if we specify any two properties of the system, then the state of the system is fully specified. In other words we can write 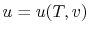 ,
, 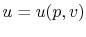 or
or 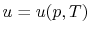 . [VW, S & B: 5.7]
. [VW, S & B: 5.7]
Consider the form , and use the chain rule to write how 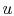 changes with respect to
changes with respect to 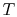 and
and 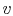 :
:
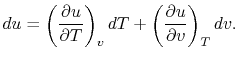 | (2..5) |
For a constant volume process, the second term is zero since there is no change in volume, 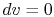 . Now if we write the First Law for a quasi-static process, with
. Now if we write the First Law for a quasi-static process, with 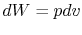 ,
, 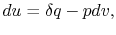 | (2..6) |
we see that again the second term is zero if the process is also constant volume. Equating (2.5) and (2.6) with 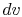 canceled in each,
canceled in each, 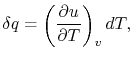 |
|
and rearranging
|
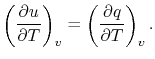 |
|
In this case, any energy increase is due only to energy transfer as heat. We can therefore use our definition of specific heat from Equation (2.4) to define the specific heat for a constant volume process,
- The Specific Heat at Constant Pressure
If we write 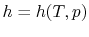 , and consider a constant pressure process, we can perform a similar derivation to the one above and show that
, and consider a constant pressure process, we can perform a similar derivation to the one above and show that
In the derivation of , we considered only a constant volume process, hence the name, ``specific heat at constant volume.'' It is more useful, however, to think of in terms of its definition as a certain partial derivative, which is a thermodynamic property, rather than as a quantity related to heat transfer in a special process. In fact, the derivatives above are defined at any point in any quasi-static process whether that process is constant volume, constant pressure, or neither. The names ``specific heat at constant volume'' and ``specific heat at constant pressure'' are therefore unfortunate misnomers; and are thermodynamic properties of a substance, and by definition depend only the state. They are extremely important values, and have been experimentally determined as a function of the thermodynamic state for an enormous number of simple compressible substances2.1.
To recap:
or
Practice Questions Throw an object from the top tier of the lecture hall to the front of the room. Estimate how much the temperature of the room has changed as a result. Start by listing what information you need to solve this problem.
The equation of state for an ideal gas is
where
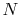
is the number of moles of gas in the volume
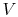
. Ideal gas behavior furnishes an extremely good approximation to the behavior of real gases for a wide variety of aerospace applications. It should be remembered, however, that describing a substance as an ideal gas constitutes
a model of the actual physical situation, and the limits of model validity must always be kept in mind.
One of the other important features of an ideal gas is that its internal energy depends only upon its temperature. (For now, this can be regarded as another aspect of the model of actual systems that the ideal gas represents, but it can be shown that this is a consequence of the form of the equation of state.) Since depends only on ,
In the above equation we have indicated that
can depend on
. Like the internal energy, the enthalpy is also only dependent on
for an ideal gas. (If
is a function of
, then, using the ideal gas equation of state,
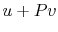
is also.) Therefore
,
If we are interested in finite changes of internal energy or enthalpy, we integrate,
and
Over small temperature changes (
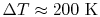
), it is often assumed that
and
are constant. Furthermore, there are wide ranges over which specific heats do not vary greatly with respect to temperature, as shown in SB&VW Figure 5.11. It is thus often useful to treat them as constant. If so
These equations are useful in calculating internal energy or enthalpy differences, but it should be remembered that they hold only if the specific heats are constant.
We can relate the specific heats of an ideal gas to its gas constant as follows. We write the first law in terms of internal energy,
and assume a quasi-static process so that we can also write it in terms of enthalpy, as in Section
2.3.4,
Equating the two first law expressions above, and assuming an ideal gas, we obtain
Combining terms,
Since
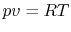
,
An expression that will appear often is the ratio of specific heats, which we will define as
Below we summarize the important results for all ideal gases, and give some values for specific types of ideal gases.
- All ideal gases:
- The specific heat at constant volume ( for a unit mass or
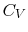 for one kmol) is a function of only.
for one kmol) is a function of only. - The specific heat at constant pressure ( for a unit mass or
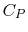 for one kmol) is a function of only.
for one kmol) is a function of only. - A relation that connects the specific heats , , and the gas constant is
where the units depend on the mass considered. For a unit mass of gas, e.g., a kilogram, and would be the specific heats for one kilogram of gas and 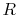 is as defined above. For one kmol of gas, the expression takes the form
is as defined above. For one kmol of gas, the expression takes the form
where and have been used to denote the specific heats for one kmol of gas and 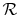 is the universal gas constant.
is the universal gas constant. - The specific heat ratio,
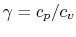 (or
(or 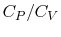 ), is a function of only and is greater than unity.
), is a function of only and is greater than unity.
- An ideal gas with specific heats independent of temperature,
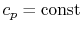 and
and 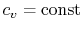 , is referred to as a perfect gas. For example, monatomic gases and diatomic gases at ordinary temperatures are considered perfect gases. To make this distinction the terminology "a perfect gas with constant specific heats" is used throughout the notes. In some textbooks perfect gases are sometimes also referred to as ideal gases, and to avoid confusion we use the stated terminology2.2.
, is referred to as a perfect gas. For example, monatomic gases and diatomic gases at ordinary temperatures are considered perfect gases. To make this distinction the terminology "a perfect gas with constant specific heats" is used throughout the notes. In some textbooks perfect gases are sometimes also referred to as ideal gases, and to avoid confusion we use the stated terminology2.2. - Monatomic gases, such as He, Ne, Ar, and most metallic vapors:
- (or ) is constant over a wide temperature range and is very nearly equal to
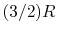 [or
[or 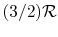 , for one kmol].
, for one kmol]. - (or ) is constant over a wide temperature range and is very nearly equal to
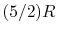 [or
[or 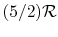 , for one kmol].
, for one kmol]. 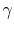 is constant over a wide temperature range and is very nearly equal to
is constant over a wide temperature range and is very nearly equal to 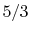 [
[ 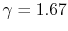 ].
].
- So-called permanent diatomic gases, namely H
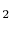 , O , N , Air, NO, and CO:
, O , N , Air, NO, and CO: - (or ) is nearly constant at ordinary temperatures, being approximately [ , for one kmol], and increases slowly at higher temperatures.
- (or ) is nearly constant at ordinary temperatures, being approximately
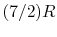 [
[ 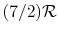 , for one kmol], and increases slowly at higher temperatures.
, for one kmol], and increases slowly at higher temperatures. - is constant over a temperature range of roughly
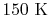 to
to 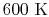 and is very nearly equal to
and is very nearly equal to 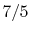 [
[ 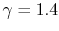 ]. It decreases with temperature above this.
]. It decreases with temperature above this.
- Polyatomic gases and gases that are chemically active, such as CO , NH
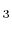 , CH
, CH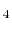 , and Freons:
, and Freons: The specific heats, and , and vary with the temperature, the variation being different for each gas. The general trend is that heavy molecular weight gases (i.e., more complex gas molecules than those listed in 2 or 3), have values of closer to unity than diatomic gases, which, as can be seen above, are closer to unity than monatomic gases. For example, values of below 1.2 are typical of Freons which have molecular weights of over one hundred.2.3
In general, for substances other than ideal gases, and 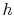 depend on pressure as well as on temperature, and the above relations will not all apply. In this respect, the ideal gas is a very special model.
depend on pressure as well as on temperature, and the above relations will not all apply. In this respect, the ideal gas is a very special model.
In summary, the specific heats are thermodynamic properties and can be used even if the processes are not constant pressure or constant volume. The simple relations between changes in energy (or enthalpy) and temperature are a consequence of the behavior of an ideal gas, specifically the dependence of the energy and enthalpy on temperature only, and are not true for more complex substances.2.4
2.4.2 Reversible adiabatic processes for an ideal gas
From the first law, with 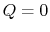 ,
, 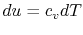 , and
, and 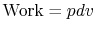 ,
,
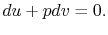 | (2..7) |
Also, using the definition of enthalpy,
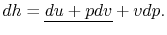 | (2..8) |
The underlined terms are zero for an adiabatic process. Rewriting (
2.7) and (
2.8),
Combining the above two equations we obtain
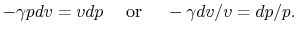 | (2..9) |
Equation (
2.9) can be integrated between states 1 and 2 to give
For an ideal gas undergoing a reversible, adiabatic process, the relation between pressure and volume is thus:
We can substitute for 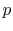 or in the above result using the ideal gas law, or carry out the derivation slightly differently, to also show that
or in the above result using the ideal gas law, or carry out the derivation slightly differently, to also show that
We will use the above equations to relate pressure and temperature to one another for quasi-static adiabatic processes (for instance, this type of process is our idealization of what happens in compressors and turbines).
Practice Questions
- On a - diagram for a closed-system sketch the thermodynamic paths that the system would follow if expanding from
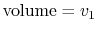 to
to 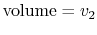 by isothermal and quasi-static, adiabatic processes.
by isothermal and quasi-static, adiabatic processes. - For which process is the most work done by the system?
- For which process is there heat exchange? Is it added or removed?
- Is the final state of the system the same after each process?
- Derive expressions for the work done by the system for each process.
Ucapan Terima Kasih:
Kepada Para Dosen di MIT dan Dosen Fisika FPMIPA Universitas Pendidikan Indonesia
Semoga Bermanfaat
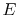 . The system energy can be considered as a sum of internal energy, kinetic energy, potential energy, and chemical energy.
. The system energy can be considered as a sum of internal energy, kinetic energy, potential energy, and chemical energy. 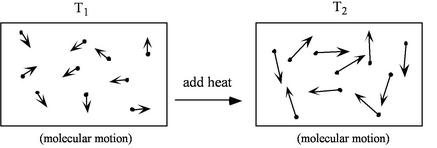
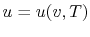 . Recall that for pure substances the entire state of the system is specified if any two properties are specified. (We will discuss the equations that relate the internal energy to these other variables as the course progresses.)
. Recall that for pure substances the entire state of the system is specified if any two properties are specified. (We will discuss the equations that relate the internal energy to these other variables as the course progresses.) 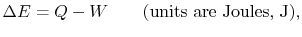
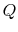 is the heat input to the system, and
is the heat input to the system, and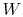 is the work done by the system.
is the work done by the system.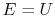 (thermal energy) +
(thermal energy) + 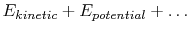
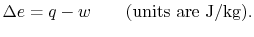
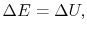
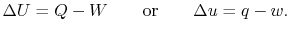
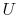 , which arises from molecular motion (see above), depends only on the state of the system;
, which arises from molecular motion (see above), depends only on the state of the system; 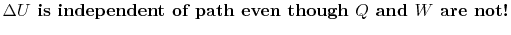
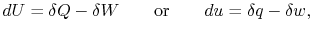
 '' is used to denote that these are not exact differentials but rather are dependent on path.
'' is used to denote that these are not exact differentials but rather are dependent on path. 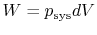 ,
,